
Count duplications and losses in the presence of polyploidy
Below are the inputs, commands, and outputs to do an analysis with GRAMPA to count the number of duplications and losses in a set of gene trees in the presence of polyploidy. The inputs are based on simulated data. For more detailed info on the simulations check our paper.
These examples were done with earlier versions of GRAMPA (<1.4.0) so some of the command line options and output formats may have changed, but the general idea and results remain the same. See the README for up-to-date info on options and formats.
Inputs
Suppose a hybridization event between the B and C lineages leading to the allopolyploid x,y,z clade has been identified. The MUL-tree representing this scenario is:
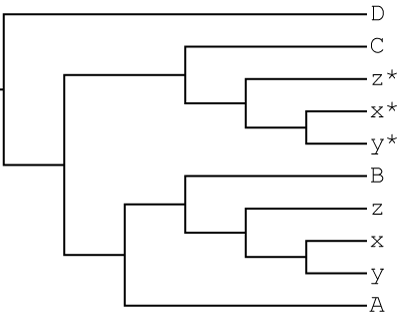
GRAMPA can accurately count duplications and losses with this MUL-tree as input.
- MUL-tree: mul_tree_74_3a.tre
- Gene trees from your set of species (in this case 1000 gene trees simulated with gain and loss) : gene_trees_3a.txt
GRAMPA command
With a known polyploidy scenario (MUL-tree), we can simply reconcile to that MUL-tree:
python grampa.py -s mul_tree_74_3a.tre -g gene_trees_3a.txt -o ex3_output -f count_test -v 0 --multreeThe --multree flag is required in this case to let GRAMPA know that the input species tree is a MUL-tree.
Outputs
The above command would create the directory ex3_output with three output files
Since we are trying to count duplications and losses, we are interested in the count_test_det.txt file. This file contains reconciliation scores for each gene tree to the lowest scoring MUL-tree (in this case, the only MUL-tree). The contents of the file look something like this:
# MT-1:((((((x*,y*)<1>,z*)<2>,B)<3>,A)<4>,(C,((x+,y+)<5>,z+)<6>)<7>)<8>,D)<9>
# GT/MT combo # dups # losses Total score
GT-1 to MT-1 5 2 7
GT-2 to MT-1 8 4 12
GT-3 to MT-1 3 0 3
GT-4 to MT-1 1 4 5
.
.
.
GT-999 to MT-1 1 2 3
GT-1000 to MT-1 2 4 6
# Gene trees with multiple maps: 36
# Total parsimony score for MT-1: 5242
The total score is reported here and in the main output file (count_test_out.txt), but with this output you can see the exact number of duplications and
losses for each gene tree, while properly counting multiple copies of the polyploid species (x,y,z) as either paralogs or homoeologs. Additionally, you
could add the --maps flag to the command above to add another column to the detailed output file that shows the maps and
duplication nodes in each gene tree (see the README section --maps for more info).