The PhyloAcc interface was run on Sunday Oct 23, 2022 at 10:03:51 EDT on bioinf02.rc.fas.harvard.edu as follows:
/n/home07/gthomas/anaconda3/envs/phyloacc-pkg/bin/phyloacc.py -d /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/seq/ -m /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/mammal_acc1.mod -t turTru2;orcOrc1;odoRosDiv1;lepWed1;triMan1 -g monDom5;sarHar1;macEug2;ornAna1 -r st -burnin 1000 -mcmc 7000 -n 24 -p 8 -j 20 -batch 25 -part holy-info,shared,holy-smokes -time 24 -o /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/mammal/st-test-branches/ -phyloacc CONSERVE_PRIOR_B 0.1;ACCE_PRIOR_A 4; ACCE_PRIOR_B 0.5; CONSERVE_PROP 0.5; CONSERVE_RATE 0.3; TRIM_GAP_PERCENT 0.7 --overwrite
| Input alignments | 2030 |
| Alignments with 0 informative sites (removed) | 1 |
| Alignments for species tree model | 2029 |
| Alignments for gene tree model | 0 |
| Batches | 82 |
| Alignments per batch | 25 |
| Processes to use per batch | 8 |
| Batches using species tree model | 82 |
| Batches using gene tree model | 0 |
| Batches to submit to cluster simultaneously | 20 |
See the full log file for more info.
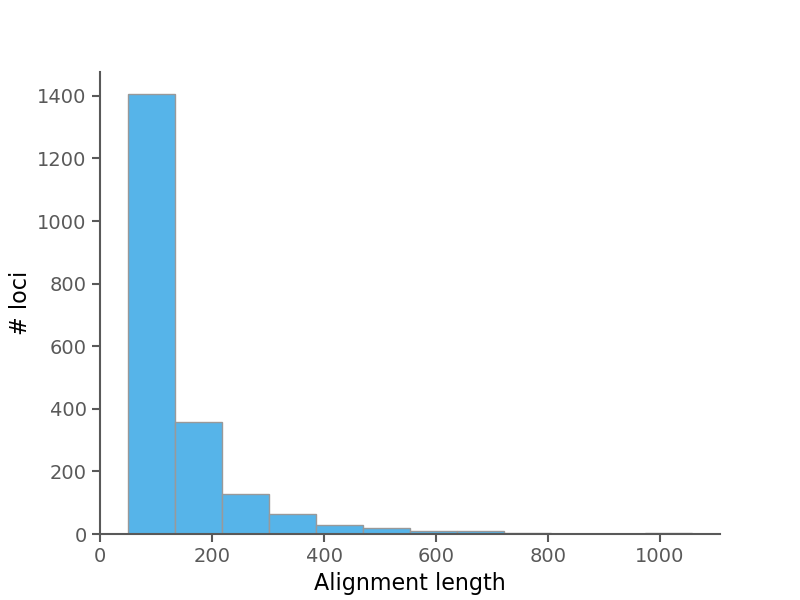
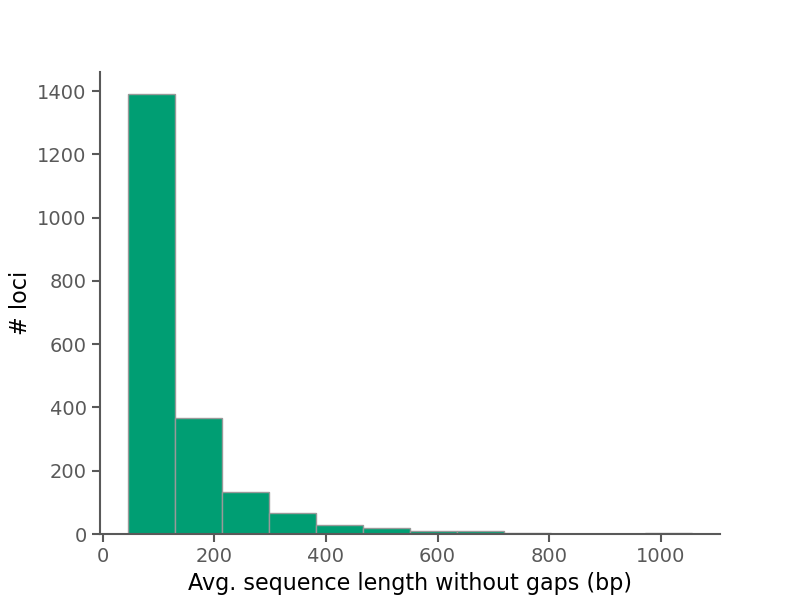
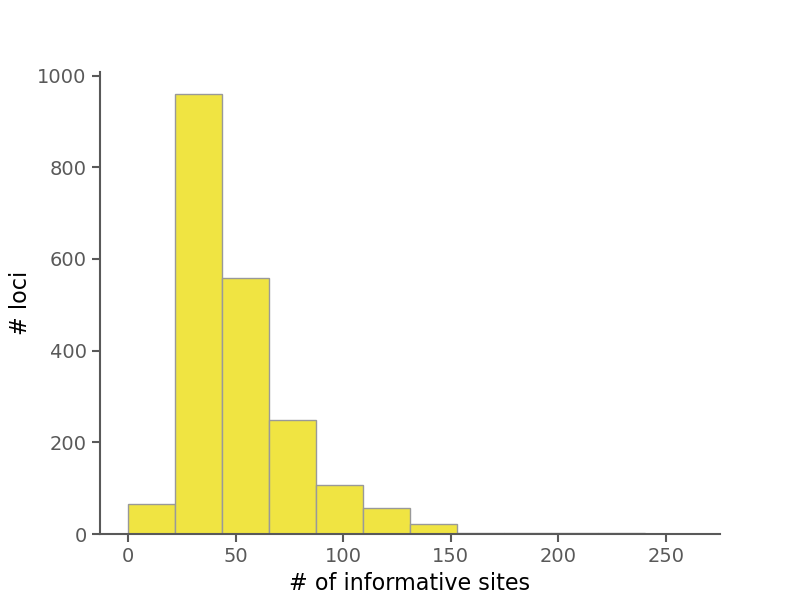
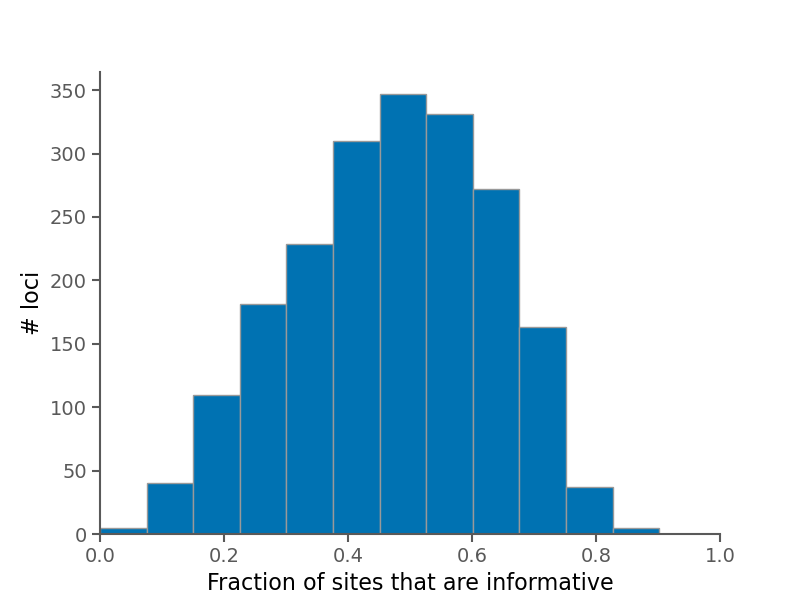
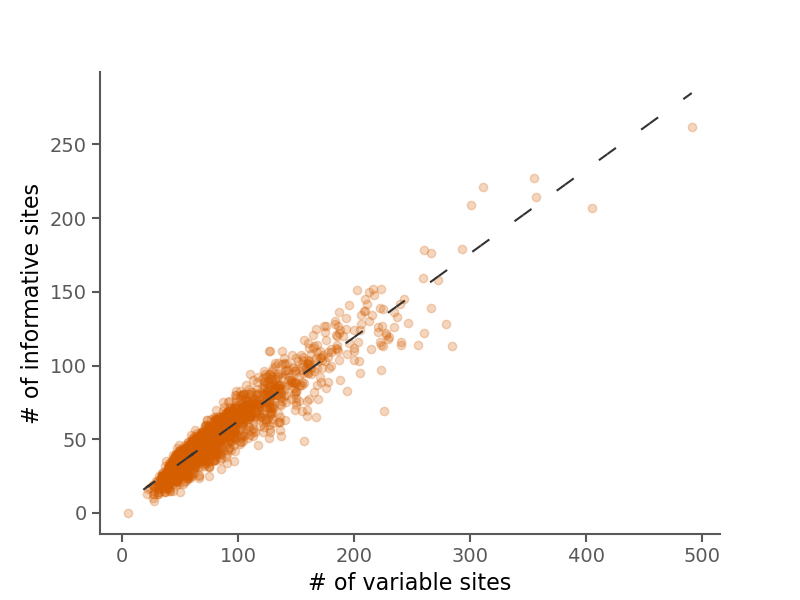
Below are some summary plots. Raw data is also available in CSV format in the alignment stats file.
The generated batches can be run with Snakemake with the following command:
snakemake -p -s /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/mammal/st-test-branches/phyloacc-job-files/snakemake/run_phyloacc.smk --configfile /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/mammal/st-test-branches/phyloacc-job-files/snakemake/phyloacc-config.yaml --profile /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/mammal/st-test-branches/phyloacc-job-files/snakemake/profiles/slurm_profile --dryrun
Note that this includes the --dryrun option to check for possible errors before submitting jobs. If everything looks good
after running that, feel free to remove the --dryrun option to submit the jobs to your cluster. You may also want to run
this in the background using screen,
tmux or some other terminal multiplexer
to avoid disruptions due to server disconnects.
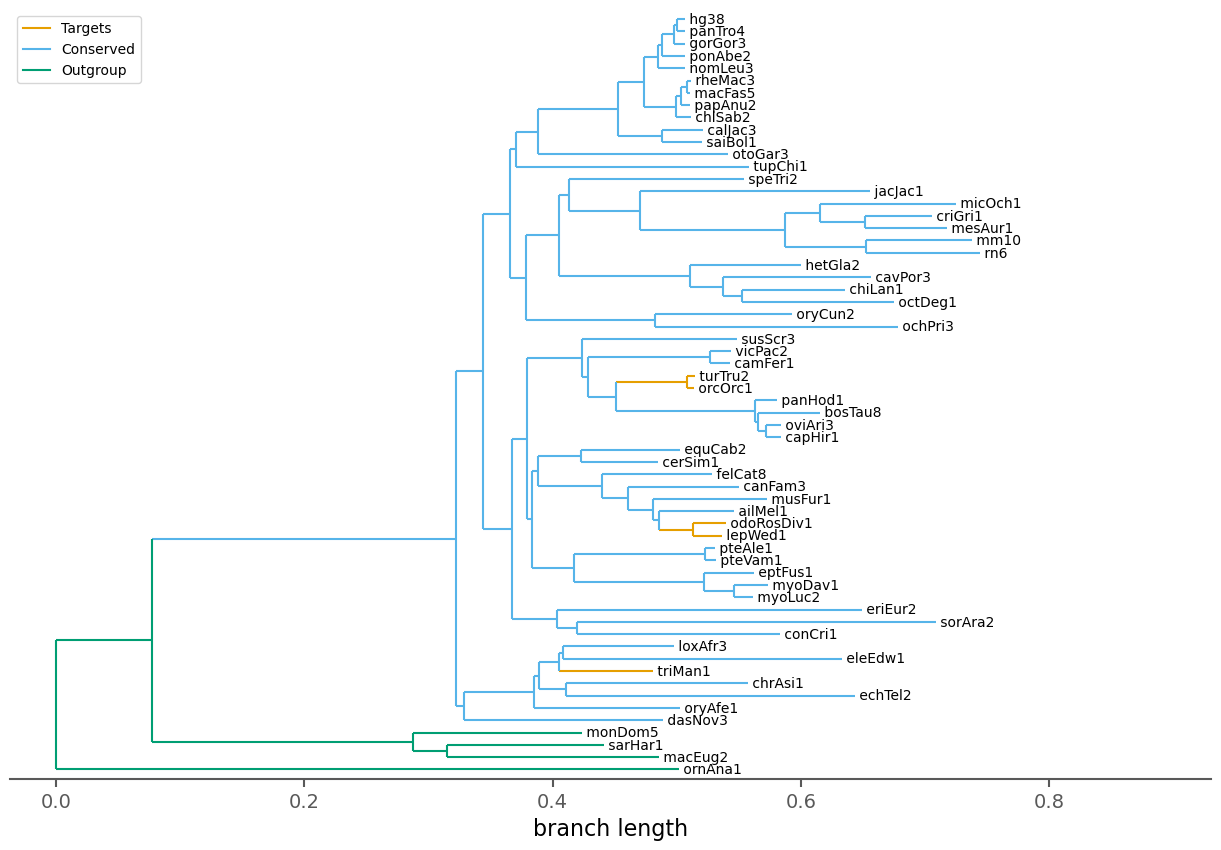
| Species | 62 |
| Target species | 5 |
| Conserved species | 53 |
| Outgroup species | 4 |
| Average alignment length | 129.627 |
| Median alignment length | 92.0 |
| Average sequence length (no gaps) per alignment | 128.71 |
| Median sequence length (no gaps) per alignment | 90.742 |