
As we described, PhyloAcc now implements a model that accounts for phylogenetic discordance in the input loci. This results in more accurate estimates of substitution rates.
Unfortunately, these models also take MUCH longer to run. That, combined with the fact that we have hundreds of thousands of loci can lead to exorbitant run times. For example, our full mammal dataset would take years to run if we ran the gene tree model on each locus one at a time.
Fortunately, we can come up with ways to speed things up:
- Not every locus may need to be run with the gene tree model, so we use site concordance factors to determine which PhyloAcc model should be used for each locus
- We batch loci to be run in parallel on the cluster with snakemake.
All that is to say that this slightly changes how we run PhyloAcc in the latest version:
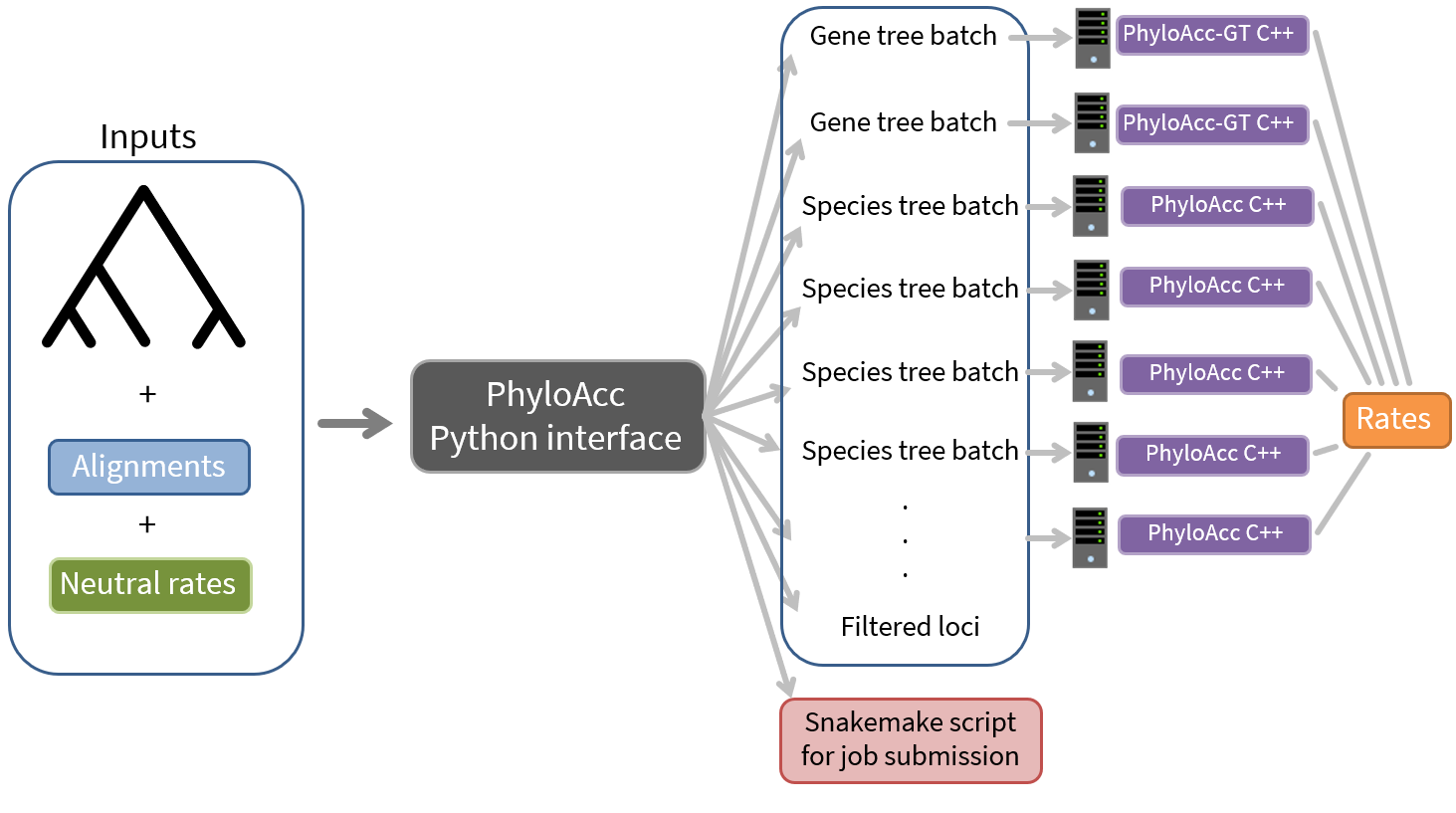
First, we will run our loci through the batching interface:
phyloacc.py -d /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/seq/ -m /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/mammal_acc1.mod -t "turTru2;orcOrc1;odoRosDiv1;lepWed1;triMan1" -g "monDom5;sarHar1;macEug2;ornAna1" -r st -burnin 1000 -mcmc 10000 -n 4 -batch 25 -part "shared" -time 4 -o phyloacc-batch-st| Command line parameter | Description |
|---|---|
| phyloacc.py | The PhyloAcc batching command |
| -d /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/seq/ | -d is the PhyloAcc option for the input directory with aligned sequences and
/n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/seq/ is the path
to that directory for our data. |
| -m /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/mammal_acc1.mod | -m is the PhyloAcc option for the input mod file with the neutral rates and phylogeny and
/n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/mammal_acc1.mod is the path
to that file for our data. |
| -t "turTru2;orcOrc1;odoRosDiv1;lepWed1;triMan1" | -t is the PhyloAcc option to set the target branches and
"turTru2;orcOrc1;odoRosDiv1;lepWed1;triMan1" are the marine mammal species in the tree. |
| -g "monDom5;sarHar1;macEug2;ornAna1" | -g is the PhyloAcc option to set the outgroup branches and
"monDom5;sarHar1;macEug2;ornAna1" are the outgroups in the mammal tree. |
| -r st | This means we want all loci to be run with the species tree model. |
| -burnin 1000 | This specifies the number of MCMC cycles to be discarded as burnin for each batch |
| -mcmc 10000 | This specifies the total number of MCMC cycles for each batch |
| -n 4 | The number of processes this script should use. |
| -batch 50 | The number of loci per batch. |
| -part "shared" | The partition on the cluster to run batches. |
| -time 4 | The time limit for each batch in hours. |
| -o phyloacc-batch-st | -o is the PhyloAcc option to specify where files are output and
phyloacc-batch-st is the path we have chosen. This directory will be created if it doesn't exist. |
This should run in about a minute, and when it's done it should have created several files and folders in the phyloacc-batch-st
directory, including a summary HTML file.
Note that all of our batches are for the species tree model. If we were to run this in adaptive mode with -r adaptive,
the program would also calculate concordance factors and run some loci with the gene tree model and some through the species tree
model based on them.
Compare the summary of an adaptive run to the species tree run above.
Mainly what this script does is partition the input loci into multiple batches and write the config files required by PhyloAcc. Let's take a look at one of those config files:
cat phyloacc-batch-st/phyloacc-job-files/cfgs/10-st.cfg| Command line parameter | Description |
|---|---|
| cat | The command to print the contents of a file to the screen. |
| /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/data/st-test-branches/phyloacc-job-files/cfgs/10-st.cfg | The path to the file you want to print. |
This should look something like this on your screen, with the paths being different based on your user name:
>PHYTREE_FILE /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/mammal-input-accelerated/mammal_acc1.mod
SEG_FILE /n/home07/gthomas/phyloacc-workshop/phyloacc-batch-st/phyloacc-job-files/bed/10-st.bed
ID_FILE /n/home07/gthomas/phyloacc-workshop/phyloacc-batch-st/phyloacc-job-files/ids/10-st.id
ALIGN_FILE /n/home07/gthomas/phyloacc-workshop/phyloacc-batch-st/phyloacc-job-files/alns/10-st.fa
RESULT_FOLDER /n/home07/gthomas/phyloacc-workshop/phyloacc-batch-st/phyloacc-job-files/phyloacc-output/10-phyloacc-st-out
PREFIX 10
BURNIN 1000
MCMC 10000
CHAIN 1
TARGETSPECIES turTru2;orcOrc1;odoRosDiv1;lepWed1;triMan1
OUTGROUP monDom5;sarHar1;macEug2;ornAna1
CONSERVE hg38;panTro4;gorGor3;ponAbe2;nomLeu3;rheMac3;macFas5;papAnu2;chlSab2;calJac3;saiBol1;otoGar3;tupChi1;speTri2;jacJac1;micOch1;criGri1;mesAur1;mm10;rn6;hetGla2;cavPor3;chiLan1;octDeg1;oryCun2;ochPri3;susScr3;vicPac2;camFer1;panHod1;bosTau8;oviAri3;capHir1;equCab2;cerSim1;felCat8;canFam3;musFur1;ailMel1;pteAle1;pteVam1;eptFus1;myoDav1;myoLuc2;eriEur2;sorAra2;conCri1;loxAfr3;eleEdw1;chrAsi1;echTel2;oryAfe1;dasNov3
NUM_THREAD 1So, this file contains paths to sequence files, mod files that have the tree and neutral rates, and a bunch of other options for the MCMC analysis
If we were performing a real analysis, we would execute the snakemake command printed out when we ran the batching script, which would submit the batches to the cluster. But since we don't have that much time, lets just run a single locus through PhyloAcc to get an idea of what its doing.
First, we'll need to create the output directory where PhyloAcc will write its files:
mkdir single-locus-output| Command line parameter | Description |
|---|---|
| mkdir | The Linux create directory command |
| single-locus-output | The name of the directory you want to create |
And then we can run PhyloAcc
PhyloAcc-ST /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/data/single-locus/1-st.cfg| Command line parameter | Description |
|---|---|
| PhyloAcc-ST | The path to the PhyloAcc species tree model program. |
| /n/holylfs05/LABS/informatics/Everyone/phyloacc-data/workshop-20221027/data/single-locus/1-st.cfg | The path to the config file for this run |
| <input file> | The path to the file you want to run on |
This should take a couple of minutes, and when its done it should create several files in the specified output directory that have information like the likelihoods of each model, the Bayes factors, and the conservation states for each branch of the tree for each locus. We'll take a closer look at all of these things when we analyze some results in a minute.
Then, after all the PhyloAcc batches have been run by the server, their outputs need to be summarized and gathered back into a single location. For this,
PhyloAcc containes a post-processing script, phyloacc_post.py, that also summarizes the results
and importantly creates the elem_lik.txt output table, which we'll analyze in detail after we
download the pre-run data...