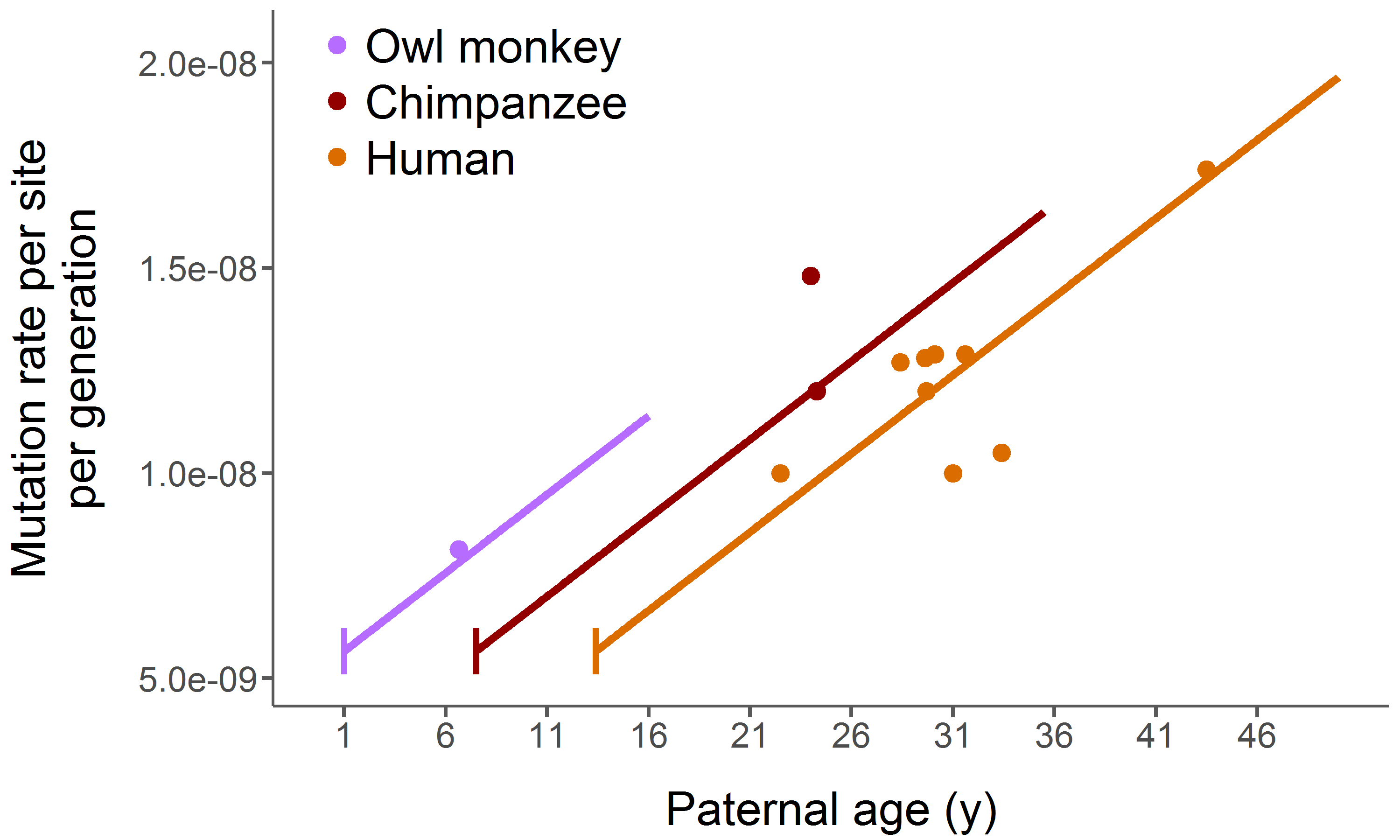
Comparing genomes between related species in the context of their phylogeny provides us the opportunity
to ask and answer questions about how these species evolved at the molecular level. Using comparative genomics
we can study patterns such as substitution rate variation, convergent molecular evolution (right, bottom), gene family
evolution (see below), gene expression and much more. I aim to uncover these patterns across the tree of
life. I have done large-scale comparative studies in primates and insects, and I am now working on similar studies
in rodents and turtles. And I am working on software to account for reference bias in read mapping.
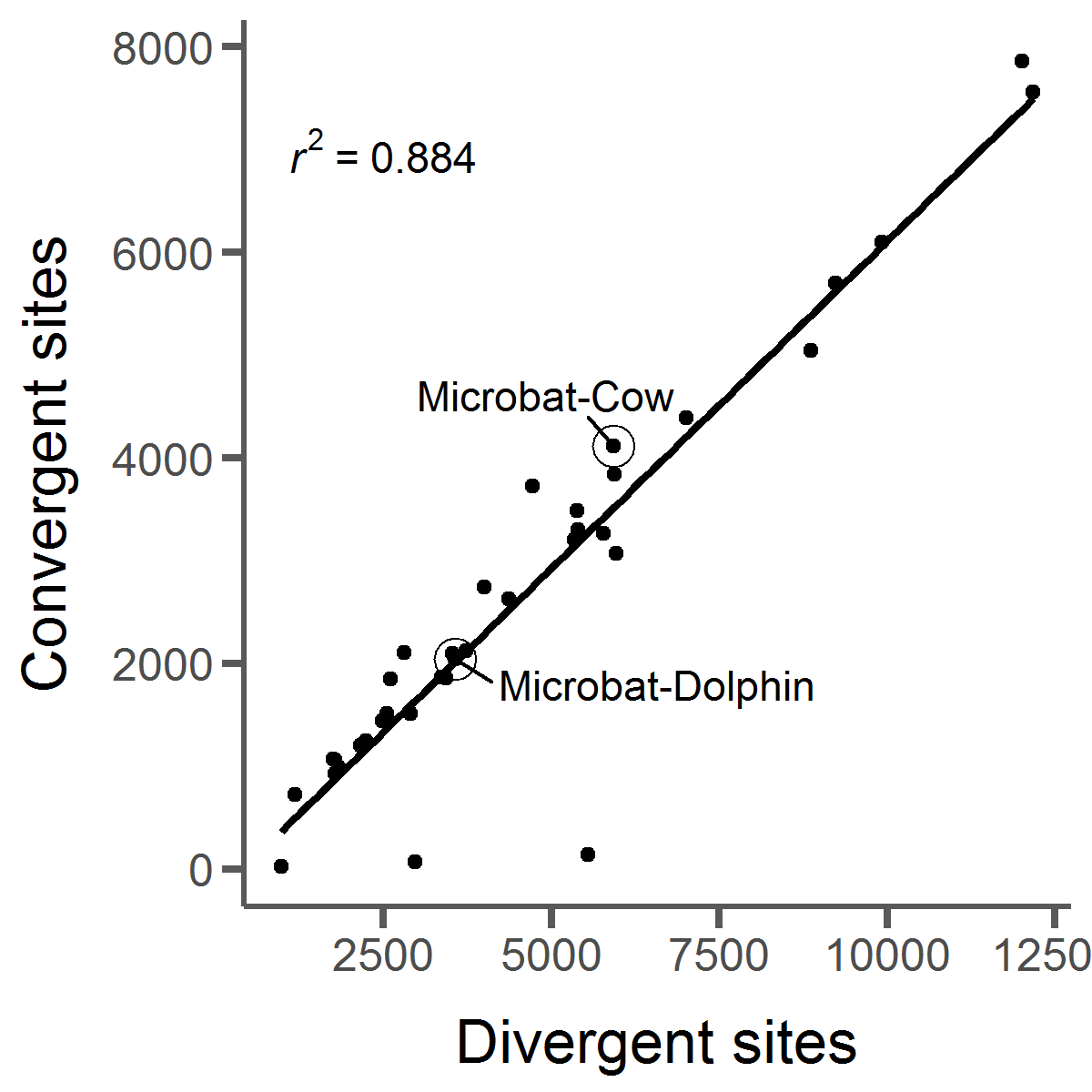
Convergent evolution occurs when distantly related lineages evolve to share the same trait. A
general assumption is that for a trait to converge in multiple lineages, it must be adaptive.
That means convergent evolution provides a great opportunity to study adaptation. Until
recently, convergent evolution was only observed at the phenotypic level (i.e. the defensive
quills on the porcupine, echidna, and hedgehog). But with whole genome sequences available for
many species we can now study convergent evolution at the molecular level and ask whether it
can be linked to convergence at the phenotypic level. However, we've discovered that convergent
amino acid substitutions occur all the time by chance in nature. This makes it difficult to pick
out which convergent changes are actually adaptive. I've done work in this area to develop
methods to detect molecular convergence while avoiding many pitfalls that come from high levels
of background convergence.
Gene duplications and losses can open up new avenues for adaptation by changing the selective
constraints and expression levels of genes. I have helped develop the latest version of
CAFE by devising a method to
estimate annotation and assembly error for gene count data. I have also delved into the study
of whole genome duplications by developing a method to properly identify and place polyploidization
events on phylogeny. This method has been implemented in the software
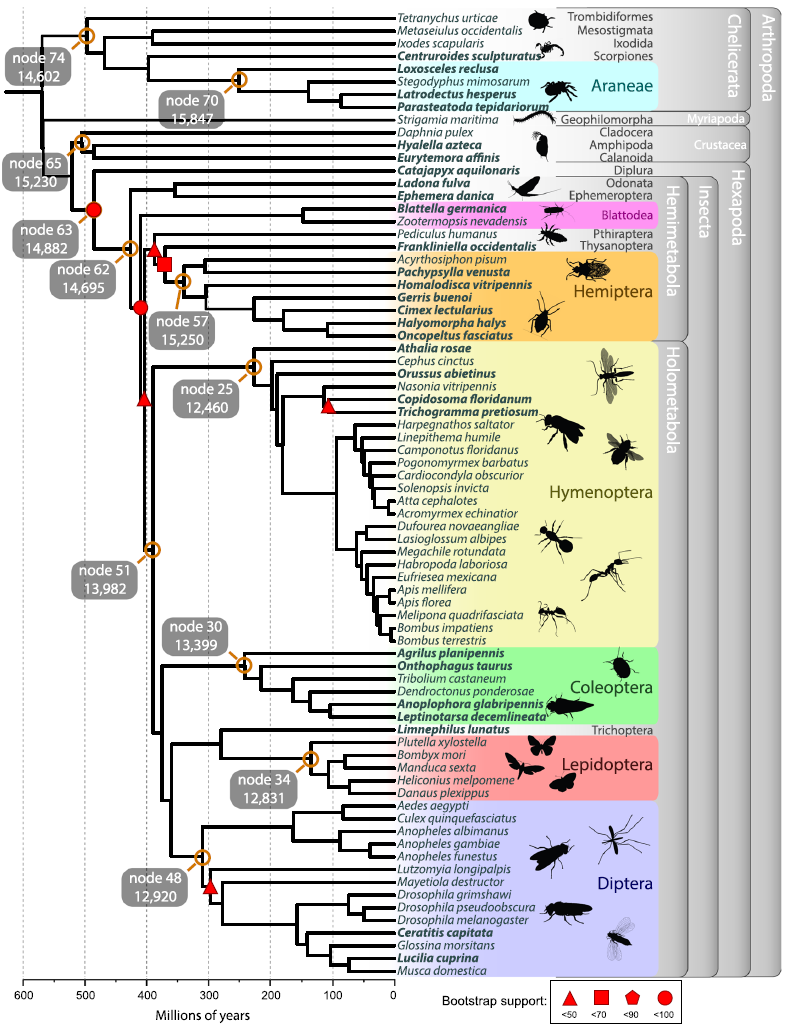
GRAMPA. Finally, I led the phylogenetic
and gene family analysis for the
i5K
pilot project (right, top). This is the largest gene family analysis to date and yielded a wealth of information as
a resource for Arthropod researchers.
A large part of comparative genomics is phylogenetics. Besides uncovering the relationships among species,
the phylogeny provides the framework for us to ask and answer many of the interesting biological questions
in comparative datasets. I am interested in studying patterns of phylogenetic discordance over time and
across the genome and developing methods to account for it in comparative analyses. Specifically, in recent
work we have found that phylogenies inferred from windows along the genome are more similar the closer they are.
This discordance poses problems when inferring changes on a single species tree: if the underlying data do not follow
that tree, evolutionary events will be mismapped. To account for this in comparative studies, I am developing methods to prune
large phylogenies to maxmize their concordance with underlying gene trees, estimate substitution rates more accurately
on a species tree, and estimating substitution rates in a Bayesian framework while accounting for discordance.