
Reference bias occurs during read mapping and causes the mapped reads to more closely resemble the reference genome than it actually does. This happens because alignment programs inherently favor alignments with a greater number of matches and with read mapping we are aligning sequences that do not match. Conceptually, this is well defined. But other than Sarver et al. 2017, little has been done to directly quantify the effects of reference bias. However, we can layout some expectations:
- Reference bias will lead to lower divergence estimates between the reference species and the mapped species.
- When mapping multiple species to a single reference, all species will appear to be more similar with respect to sequence.
- Reference bias can lead to false negatives in downstream analyses.
Briefly, reference bias occurs because some reads remain unmapped during the read mapping process. This can happen because they contain many sequencing errors (red below) OR because they contain many actual variants (green below):
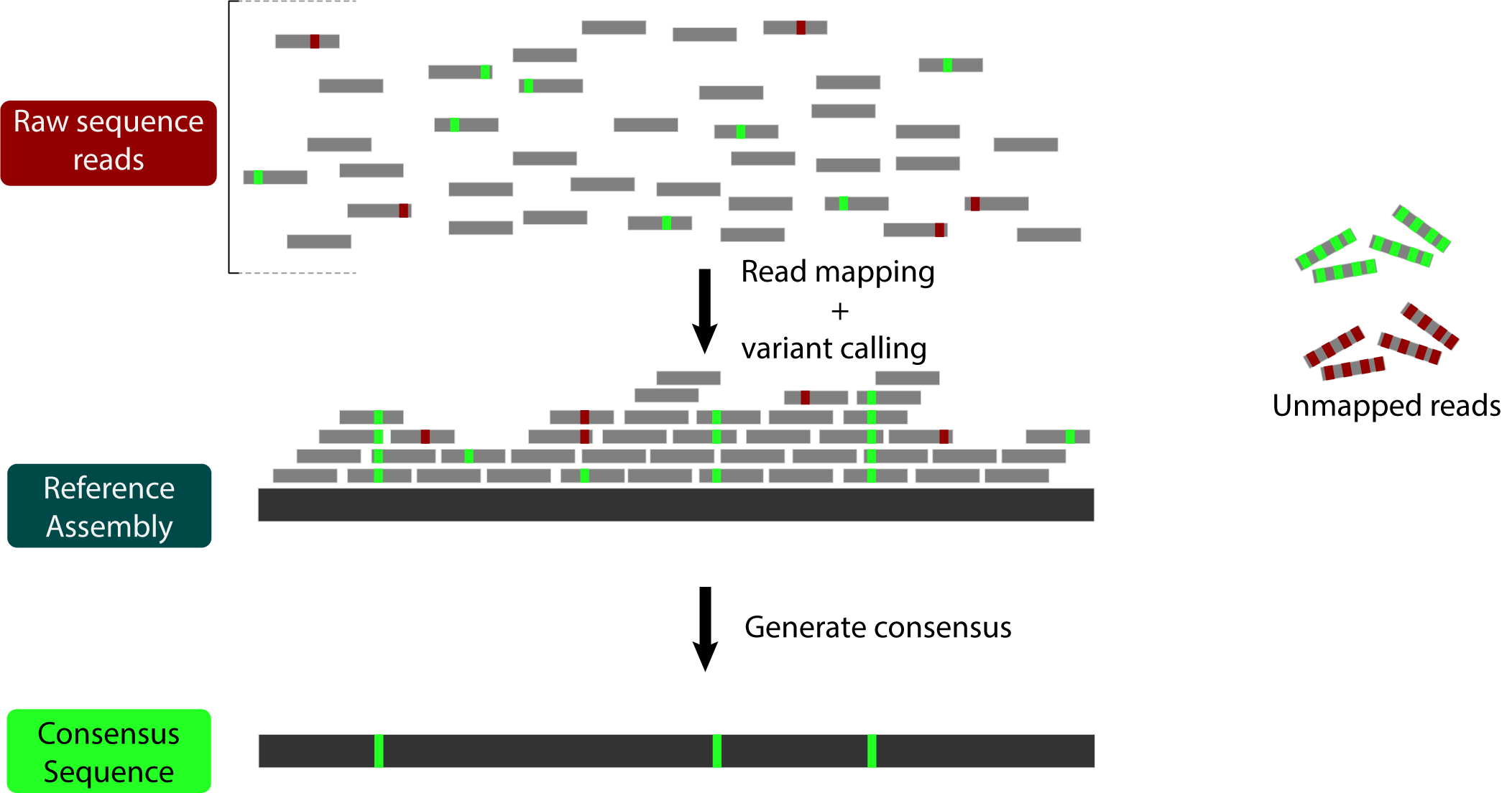
All of those unmapped reads with variants lead to our final consensus sequence looking more like the reference than it should.
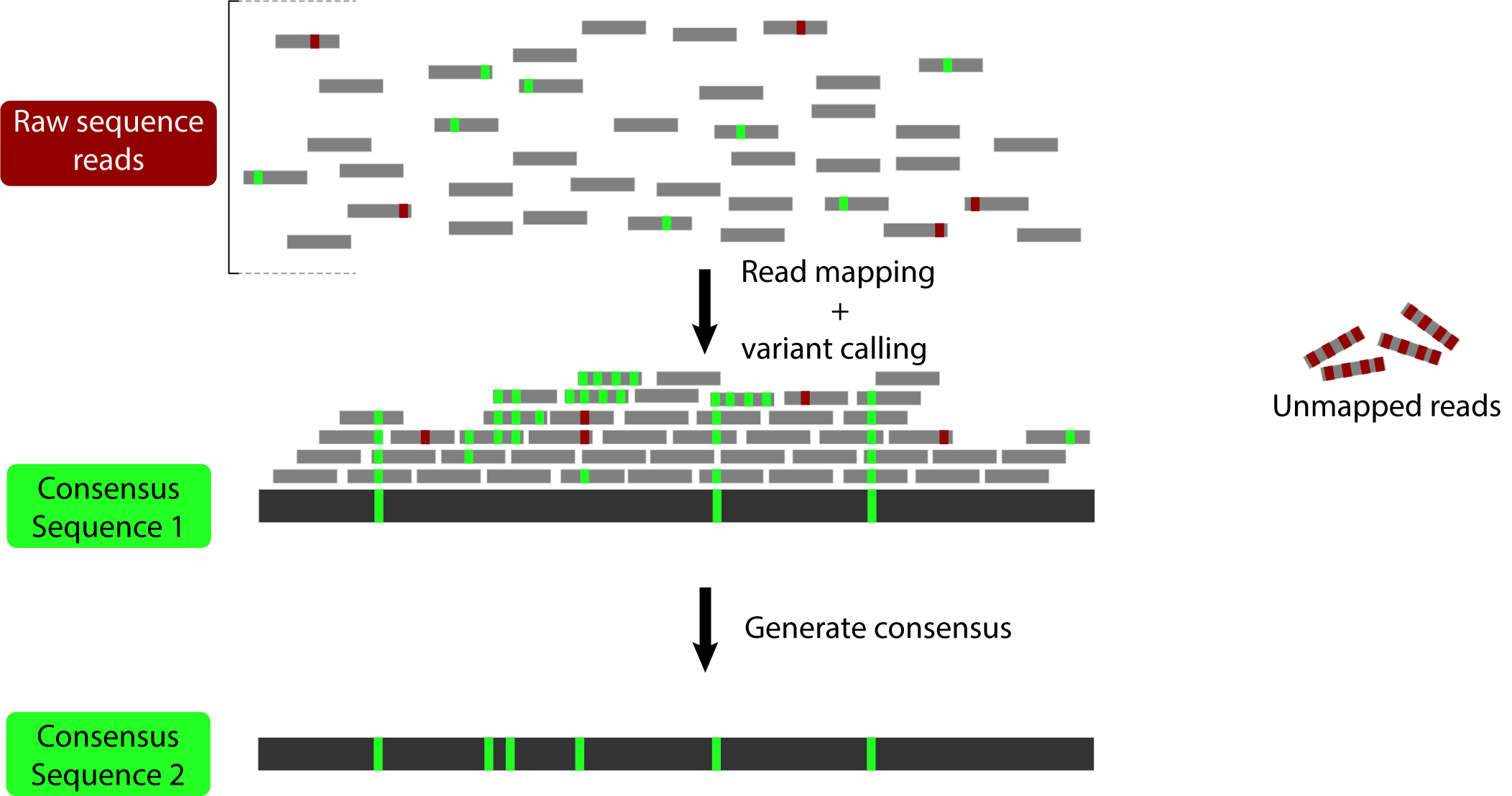
Pseudo-it is software designed to mitigate the effects of reference bias by using an iterative mapping approach to generate pseudo-reference assemblies. It performs the following steps:
- Map reads to a reference genome.
- Call variants based on the mapped reads.
- Generate a consensus sequence using the called variants.
- Repeat the process, but instead of mapping reads to the reference, map them to the newly generated consensus sequence.
The idea here is to generate a sequence that incorporates the variation that has been mapped to allow mapping of the reads that were previously too divergent:
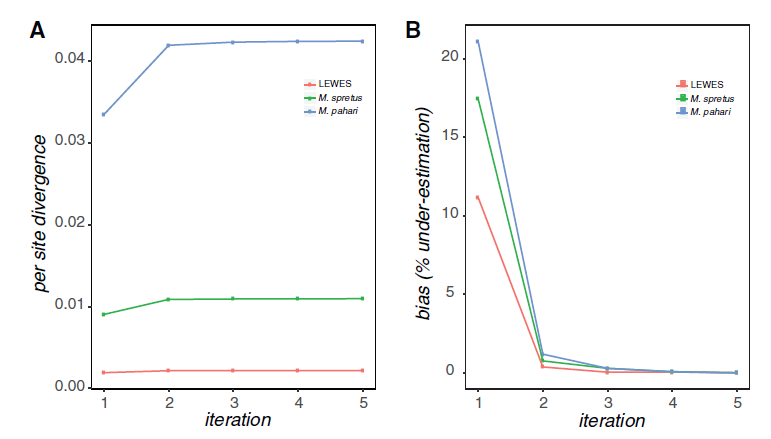
The first version of this software (Sarver et al. 2017) used BWA to map reads and GATK 3. to call variants. It was shown to increase estimated divergence in a set of mouse exomes:
The newest version that we'll be using today will still use BWA, but is upgraded to use GATK 4 and incorporates indels in the final pseudo-assembly. Let's try it out!
First, let's navigate back to the main project directory:
cd ..| Command line parameter | Description |
|---|---|
| cd | The Linux change directory command. |
| .. | The path to the directory you want to change to. .. is a shortcut that means "one directory up". |
And now we can run psuedo-it on D. pseudoobscura chromosome 2 with two iterations:
pseudo_it -ref dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa -pe1 dpse-chr2-reads/illumina/pse-chr2_1.fastq.gz -pe2 dpse-chr2-reads/illumina/pse-chr2_2.fastq.gz -i 2 -p 4 -o dpse-pseudo-it| Command line parameter | Description |
|---|---|
| pseudo_it | Call the pseudo_it program. |
| -ref dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa | Tells pseudo_it the path to the
reference genome to which we are mapping. In this case, we'll be mapping to D. melanogaster chromosome 3R. |
| -pe1 dpse-chr2-reads/illumina/pse-chr2_1.fastq.gz | Tells pseudo_it the path to the file containing the first pairs of reads. |
| -pe2 dpse-chr2-reads/illumina/pse-chr2_2.fastq.gz | Tells pseudo_it the path to the file containing the second pairs of reads. |
| -i 2 | This option sets pseudo_it to run 2 iterations. |
| -p 4 | This option tells pseudo_it to use 4 cores when mapping and calling variants.
For a whole genome, I would recommend 4 cores per chromosomes |
| -o dpse-pseudo-it | This tells pseudo_it the desired output directory. This directory will be created if it does not exist. |
This will perform the steps outlined above twice (mapping, variant calling, consensus) and will save the relevant files for each iteration, including a final consensus FASTA file as
a pseudo-assembly. You can explore the output folder dpse-pesudo-it to see what's there. You can view its progress on the screen as it runs.
Note that this runs an entire gamut of mapping, variant calling, and consensus sequence generation, which can take a very long time (about 1 week for a mammalian whole genome with 4 iterations
and 4 cores per chromosome). Even for our single chromosome from D. pseudoobscura this would take over an hour. So let's go ahead and cancel this run by hitting
<ctrl>+c while the Terminal is selected.
Sometimes you'll start a command and realize something is wrong or decide you want to do something else, so you'll want to cancel the command. In a bash terminal this is done with the following keystroke:
<ctrl>+cBut be careful not to stop a program you want to keep running!
In order to see how pseudo-it has reduced reference bias, we would first need to generate the consensus sequence after one iteration:
pseudo_it -ref dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa -pe1 dpse-chr2-reads/illumina/pse-chr2_1.fastq.gz -pe2 dpse-chr2-reads/illumina/pse-chr2_2.fastq.gz -i 1 -p 4 -resume dpse-pseudo-it| Command line parameter | Description |
|---|---|
| pseudo_it | Call the pseudo_it program. |
| -ref dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa | Tells pseudo_it the path to the
reference genome to which we are mapping. In this case, we'll be mapping to D. melanogaster chromosome 3R. |
| -pe1 dpse-2-reads/illumina/pse-chr2_1.fastq.gz | Tells pseudo_it the path to the file containing the first pairs of reads. |
| -pe2 dpse-2-reads/illumina/pse-chr2_2.fastq.gz | Tells pseudo_it the path to the file containing the second pairs of reads. |
| -i 1 | This option sets pseudo_it to run 1 iteration. |
| -p 4 | This option tells pseudo_it to use 4 cores when mapping and calling variants.
For a whole genome, I would recommend 4 cores per chromosomes |
| -resume dpse-pseudo-it | Using the resume flag tells pseudo_it that some of the files have already been generated in this
directory and it will try to resume the pipeline. In this case, we've already mapped reads for the first iteration so it shouldn't need to do that again. |
However, since this would also take some time, we'll just move forward with the provided expected output in dpse-pseudo-it.
Remember, reference bias makes mapped reads look more similar to the reference that expected, so we can outline a couple of expectations to test:
- More reads should map to the second iteration.
- The consensus sequence from the second iteration should appear to be more diverged.
01. More reads should map to the second iteration
This is fairly easy to check with samtools flagstat. First, we can run flagstat on the BAM file from the first iteration:
samtools flagstat expected-outputs/dpse-pseudo-it/iter-01/bam/merged-rg-mkdup-iter-01.bam.gz| Command line parameter | Description |
|---|---|
| samtools | Call the samtools program. |
| flagstat | Use the flagstat sub-program implemented within samtools. |
| expected-outputs/dpse-pseudo-it/iter-01/bam/merged-rg-mkdup-iter-01.bam.gz | The path to the input BAM file. |
This results in the following output:
404438 + 0 mapped (37.53% : N/A)Now let's check the second iteration:
samtools flagstat expected-outputs/dpse-pseudo-it/iter-02/bam/merged-rg-mkdup-iter-02.bam.gz| Command line parameter | Description |
|---|---|
| samtools | Call the samtools program. |
| flagstat | Use the flagstat sub-program implemented within samtools. |
| expected-outputs/dpse-pseudo-it/iter-02/bam/merged-rg-mkdup-iter-02.bam.gz | The path to the input BAM file. |
420425 + 0 mapped (38.97% : N/A)A modest increase in this example, but an increase nonetheless.
Another effect of more reads mapping is that we should have higher average read depth per iteration. Recall that with our original mappings (one iteration), we calculated an average read depth on this chromosome of 3.95, even though our expected coverage based on the Lander/Waterman equation was about 5. Let's see if our iterative mapping has improved read depth.
Then, like before, let's run samtools depth on our BAM file:
samtools depth expected-outputs/dpse-pseudo-it/iter-02/bam/merged-rg-mkdup-iter-02.bam.gz > dpse2-to-dmel3R-pi2-depth.tab| Command line parameter | Description |
|---|---|
| samtools | Call the samtools program. |
| depth | Use the depth sub-program within samtools. |
| expected-outputs/dpse-pseudo-it/iter-02/bam/merged-rg-mkdup-iter-02.bam.gz | The input BAM/SAM file. |
| > | This redirects the output that would have been printed to the screen to a file instead. |
| dpse2-to-dmel3R-pi2-depth.tab | The desired output file |
And let's calculate average depth with our custom R script:
Rscript scripts/depth_plot.r dpse2-to-dmel3R-pi2-depth.tab| Command line parameter | Description |
|---|---|
| Rscript | Invokes R to run the script. |
| ../scripts/depth_plot.r | The path to the script. |
| dpse2-to-dmel3R-pi2-depth.tab | The input file. |
This will generate a read depth histogram (depth-hist.png) and tell us the average read depth:
# Avg. read depth: 4.174893Again, a modest increase from 3.95, but a step in the right direction, showing that iterative mapping improves read depth.
02. The consensus sequence from the second iteration should appear to be more diverged.
There are many ways to calculate divergence. We'll use the simplest one: the percent identity, or the number of sites that match between the alignment of two sequences. To do that we'll first need to generate some alignments.
To generate alignments between these two chromosomes, we'll actually be using a long-read mapper called minimap2. I've found this program useful for generating between species chromosome alignments.
Next, let's use minimap2 to generate an alignment between the first iteration of mapping and the original D. melanogaster 3R reference:
minimap2 -x asm10 dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa expected-outputs/dpse-pseudo-it/iter-01/fa/iter-01-final.fa > alignments/dpse-2-pi-1-to-dmel3.paf| Command line parameter | Description |
|---|---|
| minimap2 | Call the minimap2 program. |
| -x asm10 | Set this to indicate assembly-to-reference mapping. |
| dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa | The path to the reference genome FASTA file. In this case, D. melanogaster chromosome 3R. |
| expected-outputs/dpse-pseudo-it/iter-01/fa/iter-01-final.fa | The path to the assembly FASTA file. In this case, the consensus sequence of D. pseudoobscura after one iteration of mapping. |
| > | This redirects the output that would have been printed to the screen to a file instead. |
| alignments/dpse-2-pi-1-to-dmel3.paf | The desired output file. |
minimap2 output can be formatted in two ways, the BAM format we are familiar with or PAF, the pairwise alignment format. I believe this format is unique to
minimap2, but is simply a tab-delimited file that contains informationo about blocks that have been aligned, such as sequence ID, start position, number of
matching bases, etc.
Let's take a look at this particular PAF file:
less -S alignments/dpse-2-pi-1-to-dmel3.paf| Command line parameter | Description |
|---|---|
| less | A Linux text file viewer (use space to scroll down, b to scroll up, and q to exit) |
| -S | Turn off line-wrapping within less (use the left and right arrow keys to scroll left and right). |
| alignments/dpse-2-pi-1-to-dmel3.paf | The path to the file you want to view |
3R 32168558 50522 32168555 + 3R 32079331 50522 32079328 30591593 32177084 60 tp:A:P cm:i:3034393 s1:i:30543658 s2:i:25089 dv:f:0.0028 rl:i:147655
3R 32168558 5 44485 + 3R 32079331 5 44485 23645 44480 60 tp:A:P cm:i:2055 s1:i:23645 s2:i:3382 dv:f:0.0000 rl:i:147655This shows that between the D. pesudoobscura pseudo-reference of chromosome 2 and the D. melanogaster reference of chromosome 3R there are two large blocks that could be confidently aligned (one block per line).
The following table explains the PAF columns explicitly:
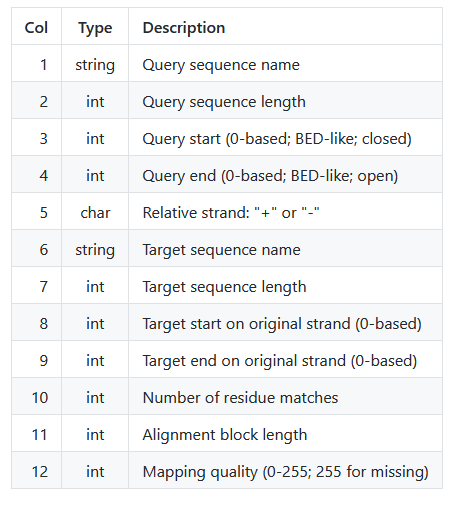
Conveniently, this tells us the number of matches (column 10) and total number of sites (column 11) per alignment block. We can use this to quickly calculate the percent identity from
a PAF file. We've provided a custom python script, div_est.py that does this. Let's run this script on the alignments/dpse-2-pi-1-to-dmel3.paf
file.
python3 scripts/div_est.py alignments/dpse-2-pi-1-to-dmel3.paf| Command line parameter | Description |
|---|---|
| python3 | Invokes python3 to run the script. |
| scripts/div_est.py | The path to the script. |
| alignments/dpse-2-pi-1-to-dmel3.paf | The input file. |
This should simply print the percent identity to the screen:
# Percent matching bases: 95.01595.015% identity after one iteration of mapping. What about after two iterations? Remember, reference bias makes mapped reads look more like the reference than expected, so we would expect % identity to DECREASE if we are improving on reference bias.
Next, run minimap2 on the second iteration of mapping:
minimap2 -x asm10 dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa expected-outputs/dpse-pseudo-it/iter-02/fa/iter-02-final.fa > alignments/dpse-2-pi-2-to-dmel3.paf| Command line parameter | Description |
|---|---|
| minimap2 | Call the minimap2 program. |
| -x asm10 | Set this to indicate assembly-to-reference mapping. |
| -x asm10 | Set this to indicate assembly-to-reference mapping. |
| dmel-3R-reference/Drosophila_melanogaster.BDGP6.28.dna.chromosome.3R.fa | The path to the reference genome FASTA file. In this case, D. melanogaster chromosome 3R. |
| expected-outputs/dpse-pseudo-it/iter-02/fa/iter-02-final.fa | The path to the assembly FASTA file. In this case, the consensus sequence of D. pseudoobscura after two iterations of mapping. |
| > | This redirects the output that would have been printed to the screen to a file instead. |
| alignments/dpse-2-pi-2-to-dmel3.paf | The desired output file. |
And then again let's calculate % identity:
python3 scripts/div_est.py alignments/dpse-2-pi-2-to-dmel3.paf| Command line parameter | Description |
|---|---|
| python3 | Invokes python3 to run the script. |
| scripts/div_est.py | The path to the script. |
| alignments/dpse-2-pi-2-to-dmel3.paf | The input file. |
# Percent matching bases: 94.541Compared to the previous 95.015%, the % identity has decreased with an iteration of mapping as expected. This means we are likely capturing more variation in this version of the pseudo-reference.