
Placing a known WGD on a phylogeny
Below are the inputs, commands, and outputs to do an analysis with GRAMPA to place a known WGD on a phylogeny. The inputs are based on simulated data. For more detailed info on the simulations check our paper.
These examples were done with earlier versions of GRAMPA (<1.4.0) so some of the command line options and output formats may have changed, but the general idea and results remain the same. See the README for up-to-date info on options and formats.
Inputs
Suppose you have a set of species, of which you have evidence that a some may be the result of a polyploidization event. You also may have an idea about the parental lineages of the polyploid species. So you build a species tree of your taxa and, since species tree reconstruction programs output singly-labeled trees, you get this as the result:
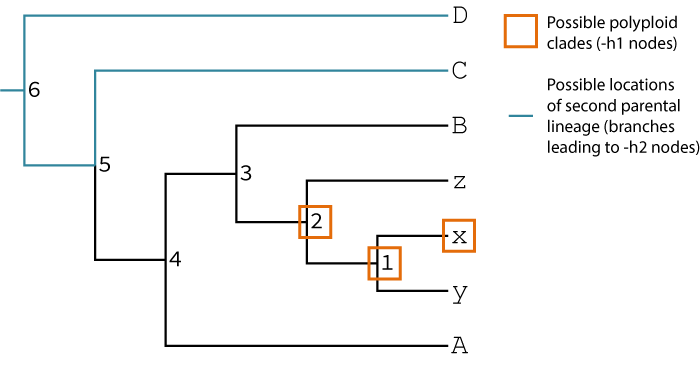
Your hypothesis is that species x, y, and z may be the result of polyploidization (but you're not sure if all of them are). The singly-labeled tree implicitly identifies one of the parental lineages of the suspected polyploids by placing the polyploid species sister to it (in this case lineage B seems to be one of the parents). You also think this tree may be the result of an allopolyploidy, and you think some lineage sister to C, D, the C,D clade, or at the root of the tree may have hybridized with some lineage related to B to form the x,y,z clade. Working with this prior knowledge GRAMPA can test your hypotheses of polyploidization.
The input files you would need are:
- Singly-labeled species tree: spec_tree_3a.tre
- Gene trees from your set of species (in this case 1000 gene trees simulated with gain and loss) : gene_trees_3a.txt
GRAMPA command
Since we have some idea of the lineages involved in the polyploidization event, we would want to limit GRAMPA's search to those lineages
with the -h1 and -h2 search parameters.
python grampa.py -s spec_tree_3a.tre -g gene_trees_3a.txt -h1 "x 1 2" -h2 "C D 5 6" -o ex2_output -f ex2_test -v 0Above we have specified -h1 and -h2 by using the node labels in the tree. Alternatively, we could specify
an equivalent -h1 and -h2 search by defining the labels based on the sets of tips that define them:
python grampa.py -s spec_tree_3a.tre -g gene_trees_3a.txt -h1 "x x,y x,y,z" -h2 "C D A,x,y,z,B,C A,x,y,z,B,C,D" -o ex2_output -f ex2_test -v 0These two commands are equivalent. The second method is slightly more cumbersome, but does not require you to have internal labels on your tree.
Although, GRAMPA can easily add internal labels to your input tree with the --labeltree command.
Outputs
The above command would create the directory ex2_output with three output files
Since we are trying to determine the mode of polyploidy, we are interested in the ex2_test_out.txt file. This file contains log info and the total reconciliation scores for each MUL-tree considered and looks something like this:
# Tree # H1 node H2 node Tree string Total score
ST ((((((x,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,D)<6> 8312
MT-1 x C ((((((x+,y)<1>,z)<2>,B)<3>,A)<4>,(C,x*)<5>)<6>,D)<7> 9109
MT-2 x D ((((((x+,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,(D,x*)<6>)<7> 8949
MT-3 x <5> (((((((x+,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,x*)<6>,D)<7> 9259
MT-4 x <6> (((((((x+,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,D)<6>,x*)<7> 9304
.
.
.
MT-11 <2> <5> (((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,C)<5>,((x*,y*)<6>,z*)<7>)<8>,D)<9> 7598
MT-12 <2> <6> (((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,C)<5>,D)<6>,((x*,y*)<7>,z*)<8>)<9> 8845
# ---------
The MUL-tree with the minimum parsimony score is MT-9: ((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,(C,((x*,y*)<5>,z*)<6>)<7>)<8>,D)<9>
Score = 5242
GRAMPA tells us MUL-tree 9 is the lowest scoring tree:
((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,(C,((x*,y*)<5>,z*)<6>)<7>)<8>,D)<9>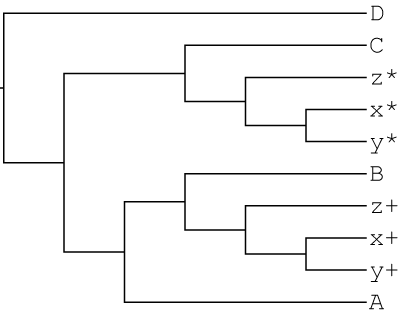
In other words, GRAMPA has identified the x,y,z clade as the polyploid clade and has identified the C lineage as the second parental lineage!