
This section will focus on analyzing SNP data from North American gray wolves (Canis lupus). The original data consist of a few hundred wolf samples that were enriched for a custom capture array consisting of several thousands of reagions across the wolf genome.
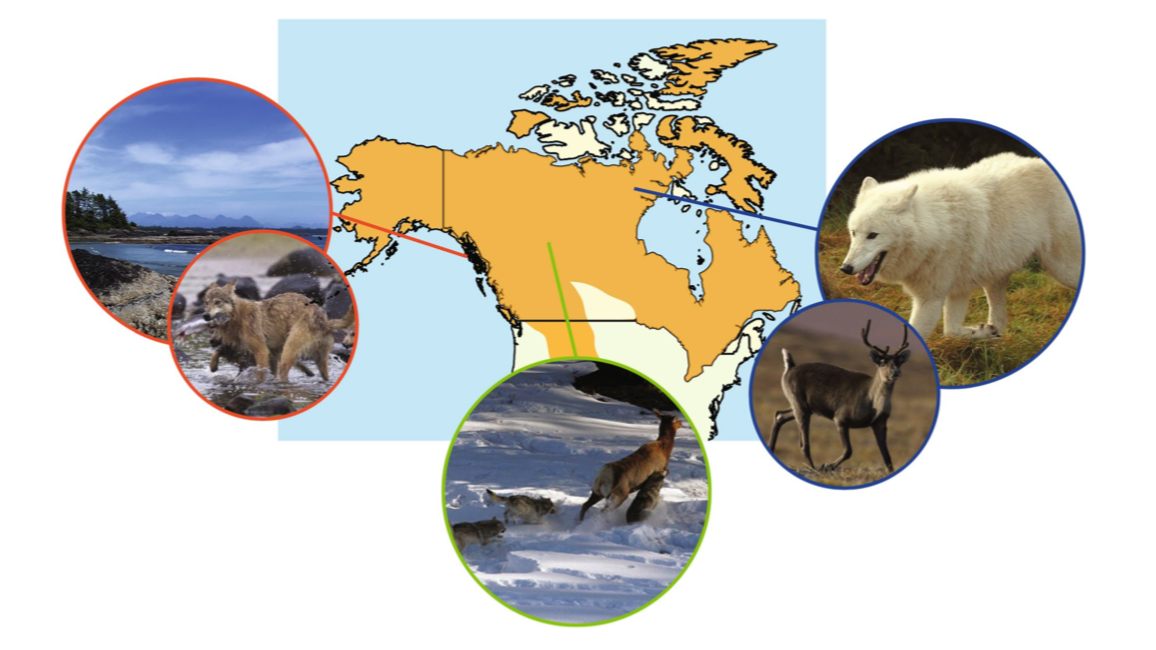
Our data consist of genic SNPs genotyped for three wolf ecotypes: (1) the British Columbia coastal, (2) the High Arctic, and (3) the Boreal Forest. These wolves have pretty striking ecological and behavioral differences that are in part driven by differences amongst their environments. We used these data to look at population genetic variation and signals of local adaptation amongst these ecotypes (Schweizer et al. 2015, Molecular Ecology).
This data is also included in the project repo you previously cloned. Let's navigate to the wolf-snps
folder now to minimize the paths we have to type when using the data. You should already be in the root project directory
(~/congen-2021-bioinformatics/), so you should just have to type the command:
cd data/wolf-snps/| Command line parameter | Description |
|---|---|
| cd | The Linux change directory command |
| data/wolf-snps | The path to the directory you want to change to. In this case,
this is a relative path. If you are not in the ~/congen-2021-bioinformatics/ directory,
you will likely get an error saying this path does not exist. |
If you are somewhere else other than the root project directory, first navigate back to that folder and then change to the
wolf-snps folder. Or, if you're familiar enough with the file tree, navigate straight to the
data folder!
Lost somewhere in the file system? Not sure exactly where you are? Remember the print working directory command:
pwd. This simply prints your current location in the file system and can help you get your
bearings if you've been moving around a lot. This and ls are essential for navigating your
file system.
I've downsampled the full data set to consist of 35 individuals and a set of genic SNPs. We will be working with these SNP data in a VCF file. VCF stands for variant call format and is just another plain text file that shows genotype data for each individual with associated quality scores, depth of coverage, etc. for each variant and sample in tab-delimited format.
The full VCF file format specifications can be found here: samtools VCF specs
VCF files alos contain a header, denoted with the # character at the beginning of the line.
The header can be a single line or thousands of lines and provides information on how the VCF file was generated, to which reference genome
the sequence data was aligned, and definitions nof the various fields in each column of the data mean. Given all this information, VCF files
can potentially be enormous in size and cumbersome to work with.
Luckily, there is software out there to help! vcftools is a suite of useful programs designed for
use on VCF formatted files. vcftools can be used to parse out SNPs and individuals, assess quality metrics of variants,
and output a variety of statistics like Fst, linkage, and heterozygosity
Much like bedtools, vcftools is desiged to follow the
Unix philosophy: commands work on properly formatted text input and and output the processed
data to the terminal, which can easily be piped (|) or redirected (>).
vcftools contains many options allowing users to perform different analyses. In general, the program is called
as follows:
vcftools <options> <input file>| Command line parameter | Description |
|---|---|
| vcftools | A program for working with variant calls in VCF format |
| <options> | Analysis specific run options |
| <input file> | The path to the file you want to run on |
Here are some more explicit examples of vcftools commands:

For this section of the workshop, we're going to start with a simple task, calculating heterozygosity, for a single wolf ecotype. Then we're going to learn how to automate it over multiple populations through using loops and writing a script.
Before we do that, let's get some basic info about out called variants in the VCF file.
TASK 1: How many SNPs are there?
We know that our VCF file has a number of header lines (which don't count towards the actual number of SNPs) and then a separate
line for each SNP. Because of this, ff we just use wc -l we will count all lines of the file, which doesn't
tell us the number of SNPs.
But by using grep with the -v flag, we can find all the lines of text that don't
contain a character, string, or pattern. Then we can use our handy | to get our answer:
grep -v "#" Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf | wc -l| Command line parameter | Description |
|---|---|
| grep | A Unix string and pattern searching command |
| -v | By default, grep prints all lines that contain
the specified string/pattern. The -v option tells grep to print
all the lines that DO NOT contain the specified string/pattern instead. |
| "#" | The string or pattern you want to search for |
| Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf | The path to the file you want to search |
| | | The pipe character means the output from the command specified before it will be used as the input tot he command specified after it |
| wc | The Linux word count command. Counts the number of lines, words, and characters in an input file. |
| -l | Tells wc to only count number of lines |
This should output the number of lines in the VCF file that don't contain the "#" character:
19620Since the only lines with "#" should be the informational header lines and all other lines represent one SNP, this means there are 19,620 SNPs represented in this VCF file.
TASK 2: Calculate heterozygosity for the coastal wolf samples.
We're going to use one of the functions of vcftools to calculate the heterozygosity
(--het) within only the coastal wolf population. vcftools
lets us specify which individuals to include in an analysis through the --keep flag with
a file of sample IDs.
vcftools --vcf Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf --out Filtered_NAwolf_n35_variableSites_GenicRegions_coastal --het --keep pop_coastal.txt| Command line parameter | Description |
|---|---|
| vcftools | A program for working with variant calls in VCF format |
| -vcf | The option to specify the input file |
| Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf | The path to the input file |
| --out | The option to specify the prefix for all output files. The file extension will be determined by the analysis. |
| Filtered_NAwolf_n35_variableSites_GenicRegions_coastal | The desired name of the output file. If it exists it will be overwritten. |
| --het | This option tells vcftools to calculate
heterozygosity of the samples in the input file. |
| --keep | This option is used with a file that lists sample IDs. Only samples in that file will be used by the program for this run. |
| pop_coastal.txt | The path to the file with the sample IDs to keep |
If we use ls to look at the output files, we can see that vcftools has provided a .log file, which contains
our command history, as well as a .het file that provides the per-individual heterozygosity.
But does this file actually give us the heterozygosity?
head Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het| Command line parameter | Description |
|---|---|
| head | A Unix command that prints the first few lines of a file |
| Filtered_NAwolf_n35_variableSites_GenicRegions_coastal | The path to the input file |
No, the output provides the number of homozygous sites and the total number of sites. But we can use this information
with a handy one-liner in awk to calculate heterozygosity:
awk '/RKW/ {print $1,$2,$3,$4,$5,(($4-$2)/$4)}' Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het > Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het.calc| Command line parameter | Description |
|---|---|
| awk | A Linux text processing language |
| '/RKW/ {print $1,$2,$3,$4,$5,(($4-$2)/$4)}' | The user-coded program to run | Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het | The path to the file on which to run the program | > | The redirect character means the output from the command specified before it will be saved to the file specified after it | Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het.calc | The path to the desired output file. This will be created if it doesn't exist and WILL OVERWRITE files that do exist |
Using this, we can also calculate the mean population heterozygosity using awk:
awk '{sum += $6} END {if (NR > 0) print sum / NR }' Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het.calc| Command line parameter | Description |
|---|---|
| awk | A Linux text processing language |
| '{sum += $6} END {if (NR > 0) print sum / NR }' | The user-coded program to run | Filtered_NAwolf_n35_variableSites_GenicRegions_coastal.het.calc | The path to the file on which to run the program |
Great! So now we have calculated the per-individual heterozygosity and the population heterozygosity for the coastal populations. Let's figure out how to repeat this set of commands for any number of additional populations.
Instead of hard-coding our command to be specific to the coastal wolves, we can use shell variables. We indicate a shell variable like so:
wolf_pop=coastal| Command line parameter | Description |
|---|---|
| wolf_pop | The desired variable name appears to the left of the equals sign |
| = | The equals sign indicates variable assignment in programming languages | coastal | The value you wish to assign to the variable name |
We can check that variable assignment was successful by printing the value with the echo command:
echo ${wolf_pop}| Command line parameter | Description |
|---|---|
| echo | The Unix print command. Values given as input will be printed to the screen. |
| ${wolf_pop} | The item to print to the screen. In this case, because wolf_pop is a
defined variable, we must use the ${} symbols to let echo know to print it's assigned value to the screen, rather
than just the text "wolf_pop". |
We can use variables in any command in the terminal. For instance, we can run the same command as before to calculate heterozygosity, but instead of typing "coastal" in the command, we can use our variable:
vcftools --vcf Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf --out Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop} --het --keep pop_${wolf_pop}.txt| Command line parameter | Description |
|---|---|
| vcftools | A program for working with variant calls in VCF format |
| -vcf | The option to specify the input file |
| Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf | The path to the input file |
| --out | The option to specify the prefix for all output files. The file extension will be determined by the analysis. |
| Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop} |
The desired name of the output file. If it exists it will be overwritten. In this case, we use the wolf_pop
variable to create a file with the name of the current population. |
| --het | This option tells vcftools to calculate
heterozygosity of the samples in the input file. |
| --keep | This option is used with a file that lists sample IDs. Only samples in that file will be used by the program for this run. |
| pop_${wolf_pop}.txt | The path to the file with the sample IDs to keep. In
this case, we're using our variable, wolf_pop to indicate which existing file to use. |
This provides the same output as the command we ran earlier. So, we could assign a new value to the wolf_pop
variable. Notice also how I have been consistent in how I named all of my pop files so that I can substitute an ecotype
name but otherwise still have that same general file name.
wolf_pop=highArctic| Command line parameter | Description |
|---|---|
| wolf_pop | The desired variable name appears to the left of the equals sign |
| = | The equals sign indicates variable assignment in programming languages | highArctic | The value you wish to assign to the variable name |
We can check that variable assignment was successful by printing the value with the echo command:
echo ${wolf_pop}| Command line parameter | Description |
|---|---|
| echo | The Unix print command. Values given as input will be printed to the screen. |
| ${wolf_pop} | The item to print to the screen. In this case, because wolf_pop is a
defined variable, we must use the ${} symbols to let echo know to print it's assigned value to the screen, rather
than just the text "wolf_pop". |
Now that we've reassigned the wolf_pop variable to the highArctic population, we can run the same series of commands as above,
but vcftools will use the samples and provide output for this population.
First, run vcftools to count the number of sites:
vcftools --vcf Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf --out Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop} --het --keep pop_${wolf_pop}.txt| Command line parameter | Description |
|---|---|
| vcftools | A program for working with variant calls in VCF format |
| -vcf | The option to specify the input file |
| Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf | The path to the input file |
| --out | The option to specify the prefix for all output files. The file extension will be determined by the analysis. |
| Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop} |
The desired name of the output file. If it exists it will be overwritten. In this case, we use the wolf_pop
variable to create a file with the name of the current population. |
| --het | This option tells vcftools to calculate
heterozygosity of the samples in the input file. |
| --keep | This option is used with a file that lists sample IDs. Only samples in that file will be used by the program for this run. |
| pop_{wolf_pop}.txt | The path to the file with the sample IDs to keep. In
this case, we're using our variable, wolf_pop to indicate which existing population file to use. |
Then run awk to calculate heterozygosity for each individual in the highArctic sample. Again note that we use the
wolf_pop variable in our input and output file names:
awk '/RKW/ {print $1,$2,$3,$4,$5,(($4-$2)/$4)}' Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het > Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc| Command line parameter | Description |
|---|---|
| awk | A Linux text processing language |
| '/RKW/ {print $1,$2,$3,$4,$5,(($4-$2)/$4)}' | The user-coded program to run | Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het |
The path to the file on which to run the program. Note that we use the wolf_popvariable to indicate
the population file to run. |
> | The redirect character means the output from the command specified before it will be saved to the file specified after it | Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc |
The path to the desired output file. This will be created if it doesn't exist and WILL OVERWRITE files that do exist.
Note that we use the wolf_popvariable to save to a file name labeled with the correct population. |
And then awk again with the wolf_pop variable to get mean population heterozygosity:
awk '{sum += $6} END {if (NR > 0) print sum / NR }' Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc| Command line parameter | Description |
|---|---|
| awk | A Linux text processing language |
| '{sum += $6} END {if (NR > 0) print sum / NR }' | The user-coded program to run | Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc |
The path to the file on which to run the program. Note that we use the wolf_pop variable to indicate
the population file to run. |
Now, we're going to add some of these commands with variables to a script so that we can automate the series of commands to generate per-individual and per-population heterozygosity.
A script is simply a text file where we can write and save the code and commands we want to run. Scripts give use the flexibility to perform complex operations based on conditions and ranges of values. Since a script is a text file, we're going to create a new text file and copy some commands into it. We're going to use an in-terminal text editor, vim to do this, although there are several other options.
vim het_analysis.sh| Command line parameter | Description |
|---|---|
| vim | An in-terminal text editor available on Linux. |
| het_analysis.sh | The name of the text file to create or view (if it exists). The .sh extension indicates this is a shell script |
You should now be in the vim text editor. Your screen should be cleared of commands and cursor should be blinking on top, with
tilde characteres (~) on every other line. Importantly, vim has both a viewing mode and an editing mode.
When you create a new file with vim, as we have here, you start out in viewing mode. We can switch to editing mode
by pressing the a key. Once you do this, you'll see the text -- INSERT -- at the bottom of the screen, indicating we have
switched to editing mode.
Now, we can copy and paste our commands for calculating heterozygosity into this file. Of course, there are other ways we could get the
commands into the file (such as using echo and redirecting), but we'll stay simple today.
Copy the following commands:
wolf_pop=$1
vcftools --vcf Filtered_NAwolf_n35_variableSites_GenicRegions.recode.vcf --out Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop} --het --keep pop_${wolf_pop}.txt
awk '/RKW/ {print $1,$2,$3,$4,$5,(($4-$2)/$4)}' Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het > Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc
awk '{sum += $6} END {if (NR > 0) print sum / NR }' Filtered_NAwolf_n35_variableSites_GenicRegions_${wolf_pop}.het.calc
The last three commands should be familiar to you already. They are the same commands we used above for the coastal population, but
we've substituted the string "coastal" for the variable wolf_pop.
The first line, wolf_pop=$1 is our variable assignment. It tells the script to assign the variable wolf_pop the value
of $1. Well what is $1? It has a dollar sign ($) preceding it, so we know it is a variable. But we haven't
assigned anything to it yet.
Like other commands, your own scripts can take input options from the command line. By default, a shell script (like the one we are
writing) will assign these options to internal variables: $1 for the first option, $2 for the second one, and so on.
So, wolf_pop will be assigned whatever we type into the terminal when we run the script.
Let's exit out of vim to try and run our script. First, switch back to viewing mode by pressing the ESC key.
You should no longer see the text -- INSERT -- at the bottom of the screen. When in viewing mode, any input we type
is interpreted as internal vim commands, such as saving, exiting, etc. Let's tell vim to save the changes we've made to
the file with the write command (w) and then quit (q) the text editor by typing:
:wq| Command line parameter | Description |
|---|---|
| : | Tells vim to interpret the following text as commands |
| w | The vim write command that saves changes made to the file |
| q | The vim quit command to exit the text editor and return to the
terminal |
TASK 3: Make the script executable and try it
We will use the chmod command to change the permissions on the script to the shell knows that it's contents
should be interpreted as commands. This is known as making the file executable:
chmod a+x het_analysis.sh| Command line parameter | Description |
|---|---|
| chmod | The Unix change mode command used to change permissions on files |
| a+x | This tells chmod to modify (a the file's permissions
to make it executable (+x) |
| het_analysis.sh | The name of the file to change permissions |
Now let's try to execute our file on the coastal population:
./het_analysis.sh coastal| Command line parameter | Description |
|---|---|
| ./het_analysis.sh | The name of the file to run. Since we have changed its
permissions to be executable, its contents will be interpreted as shell commands. ./ also indicates to
run this file as a shell script |
| coastal | Remember, the script we wrote expects input from the command
line to assign to the $1 variable. This will be that value |
If we use our handy ls -lht command, we can see that the three newest files are of the coastal wolf samples, and that
the script we wrote printed the mean population heterozygosity to the screen.
Documenting our code with comments and print statements
It is also good practice to document what each step of our code does in human-readable language. This is called commenting
our code. The way to comment code differs between programming languages, but a fairly common method (used in Python, R, and the shell
scripting we're doing) is with the # symbol. For languages that use this symbol, any line starting with # will
be interpreted as a comment rather than a command to be run.
We can also provide some helpful context to the output of our script. Rather than just printing out a number that may be confusing
to someone else using the script (or even ourselves if we re-run this same script months or years later), we can use echo
within the script to print some more information, like that the number printed represents mean population heterozygosity.
Feel free to open up your script and follow along as I comment my version and add contextutal print statements:
vim het_analysis.sh| Command line parameter | Description |
|---|---|
| vim | An in-terminal text editor available on Linux. |
| het_analysis.sh | The name of the text file to create or view (if it exists). The .sh extension indicates this is a shell script |
TASK 4: Which population has the highest mean heterozygosity?
To find this out now, all we have to do is run our script on all three populations:
./het_analysis.sh coastal
./het_analysis.sh highArctic
./het_analysis.sh borealForestThis shows that the High Arctic populations has the highest mean heterozygosity at 0.36179.
for loops
If only there was a way to automate the analysis even further... Aha! We can use for loops to call the script
multiple times with different input values, in this case our 3 population strings, for the wolf_pop variable.
for loops in a shell script are generated in a similar way as in other programming languages:
for <variable> in <iterable>; do <code>; done| Command line parameter | Description |
|---|---|
| for | The start of the for loop |
| <variable> | for loops iterate over a set of values. Whatever
is named here as |
| in | Indicates that the following will be the set of values to iterate over |
| <iterable>; | A data type that contains multiple values |
| do | A keyword that indicates a block of code within a for loop
has started |
| <code>; | The code to execute on each value in the set of values |
| done | A keyword that indicates the end of the for loop |
For instance, we could do the following to loop over letters in the alphabet from A to F and print them out:
for letter in {A..F}; do echo $letter; doneOr the following to subtract 1 from each integer from 1 to 4 and print the resulting value to the screen:
for num in 1 2 3 4; do echo "The value of $num - 1 is " `expr $num - 1`; doneTASK 5: Use a for loop to calculate heterozygosity for the three wolf ecotypes
for pop in coastal highArctic borealForest; do ./het_analysis.sh $pop; done| Command line parameter | Description |
|---|---|
| for | The start of the for loop |
| pop | This tells the for loop to assign the current value
being executed to the $pop variable |
| in | Indicates that the following will be the set of values to iterate over |
| coastal highArctic borealForest | The values over which to iterate |
| do | A keyword that indicates a block of code within a for loop
has started |
| ./het_analysis.sh $pop; | Run the het_analysis script on the current value
stored in $pop |
| done | A keyword that indicates the end of the for loop |
This should produce the same output as when we ran the three commands manually above!
Hopefully you've become more familiar with how to use variables in the command-line and in scripts, how to write a script and edit it
using vim, and how to use for loops to make your life easier!